Estimating Pathogenic Probability
You are here!

The haploid human genome consists of over three billion individual units of information (nucleotides or base pairs) and for a simple Mendelian disease, even a single variation at one of these three billion sites could be responsible for a patient’s disease. At each nucleotide position, there are four choices: A, T, C, and G. If every nucleotide in the haploid human genome were the size of a common penny, three billion of them placed side-by-side along the equator would circle the Earth 1.5 times. Assuming that these pennies were all minted during a four-year period, finding the single nucleotide change responsible for this patient's disease would be the same as circling the globe at the equator one and a half times looking for a variation in the year on one of the pennies. To make things even more challenging, there is an enormous amount of "normal" variation in the genomic sequence of human beings. Any two random individuals will differ from one another at at least one of every 1000 nucleotides [12]. Thus, even if it were technically possible to compare the entire genomic sequence of a given patient to that of a "normal" individual, the test would reveal at least three million differences, any single one of which could potentially cause the patient's disease. To put it another way, any single variation from the "normal" genomic sequence that one might detect in a patient with a genetic disease has less than a one in a million chance of being responsible for that patient’s disease unless it has been previously demonstrated to be significantly associated with the disease in a carefully controlled experiment with a large number of patients.
Sequence Variations and Phenotypes
There are two ways that a sequence variation in the genome can be meaningfully related to an alteration in the structure or function of the person that harbors it. The most obvious way is that it can cause the altered phenotype by affecting the function of one or more genes in a significant way. The other is that it can be tightly linked (i.e., on the same chromosome and so close that it is unlikely to be separated by a recombination event) to some other sequence variation that is causally related to the phenotype. For some clinically relevant purposes (such as carrier testing in a family affected with an X-linked disease), it does not matter whether a variant causes the change in the phenotype or is very tightly linked to one that does. In other cases (such as trying to deduce the function of a specific domain of a protein by characterizing the effect of a variation within that domain) it does matter quite a bit. Some kinds of experimental data will support the idea that a variant does alter the function of a gene, while other kinds of data speak only to its physical association with a gene whose function is altered.
Of course, most sequence variations will have no relationship to a patient's phenotype at all. For clinicians trying to use genomic data to help care for their patients, it is helpful to have a system for estimating (and communicating) in a standardized fashion the likelihood that a sequence variation is related to a patient's disease, especially if both functional and association information can be combined in a readily understandable way.
Estimate of Pathogenic Probability (EPP)
The system used by the Carver Laboratory combines all readily available functional and association information into a score known as the Estimate of Pathogenic Probability (EPP). This system is applicable only to variations in genes that have already been statistically proven to be associated with a given phenotype. But, it is useful for helping clinicians decide whether a variation is likely to be responsible for a disease that an individual already manifests. It is also useful for ranking members of a group of individuals who harbor sequence variations in a given gene according to the likelihood that their disease is caused by that gene. This may in turn be useful for selecting individuals who would be most likely to benefit from gene-replacement therapy or for selecting which individuals' clinical data to sum when trying to determine the "natural history" of a given disease. The EPP system provides an objective set of rules for communicating all that is known about the pathogenic probability of a given variant. It does not require hours of deliberation among highly trained people (which could introduce all sorts of unpredictable personal bias) and it can be easily revised as new data become available. Both of these latter features are highly desirable when one considers the volume of genotype/phenotype data that is accumulating in even a single field like ophthalmology (as evidenced, for example, by the mutation data summarized on this website).
It would be ideal if a system for estimating pathogenic potential could be totally mathematical and have every term in the calculation rigorously supported by well-established statistical theory and large data sets. We must admit that the EPP system that we currently use does not meet this standard and is more of a first approximation that we hope will serve as a precursor to a more sophisticated system in the future. However, we have given quite a bit of thought to objective methods for capturing as much information as possible from the structure of the gene itself and from the way that the gene’s alleles are distributed among patient and control groups. However, some of the decisions regarding the weights that various factors are given in the final EPP score were not derived mathematically but were determined empirically based upon our experience in analyzing real families with mutations in the genes that are tabulated on this website.
There are currently two sets of rules and two sets of interpretations for EPP values: one for autosomal dominant conditions and one for autosomal recessive ones. The methods are applicable to X-linked diseases as well, but we have not yet devised the specific empirical strategy for weighting the various factors for this inheritance pattern nor are our data for any X-linked conditions included in the Appendix. In all cases, the EPP has four possible values: 0, 1, 2, and 3. An EPP of 0 means that a variation has very little probability of causing or being meaningfully associated with a disease while an EPP of 3 means that it is extremely likely that a variation is responsible for the disease. Values of 1 and 2 indicate intermediate likelihoods that a variant is responsible for a patient’s disease and have slightly different interpretations depending on whether the disorder in question is autosomal dominant or recessive. The dominant case is the simplest: the higher numbers simply reflect higher pathogenic potential. With recessive disease, one has to consider the possibility that all alleles do not contribute equally to a recessive phenotype. That is, there may be “low penetrance” alleles that are too common in the population for them to be involved in classic recessive inheritance. This subject is discussed in detail in our paper on allelic variation in the ABCA4 gene [13], and to date, it is the ABCA4 gene that benefits the most from this additional nuance in the EPP interpretation. For recessive diseases like Stargardt disease, an EPP of 1 indicates a "possible low penetrance allele" while an EPP of 2 indicates a "possible highly penetrant allele" and an EPP of 3 indicates a "probable highly penetrant allele."
Calculating the EPP
The EPP is calculated using all readily available information about the function of the variant allele and its previous association with disease. Before considering the details of the calculation of EPP, it may be helpful to consider the types of functional and association information that might be used for such a calculation and the practical limitations of each.
Functional data used in the calculation
The most obvious way that one could assess the functional effect of a given sequence alteration would be to sample (e.g. biopsy) tissue expressing the variant protein and measure the function of the protein directly. When available, this type of information is the most reliable and would obviate the need for an EPP-type system. This approach has been used widely in medicine, especially in the pre-genomic era. The demonstration of the functional defect in beta-globin in patients with sickle cell disease would be an example of this approach. Unfortunately, this is rarely possible in ophthalmology for two reasons. First, most affected tissues of the eye are not amenable to biopsy from living individuals, and second, the function of most newly discovered genes is so poorly understood that a meaningful assay of their function does not exist even if the tissues were available.
A second, related approach for investigating the function of an altered allele is to create an animal or in vitro model of a disease by artificially expressing (or inhibiting) the gene experimentally. In general, this is a powerful approach but is not without limitations. The main limitation is that the more closely the experiment matches the human situation, the more expensive it is and hence the less practical for assessing hundreds of sequence variations. The less the experiment matches the human situation, the more one has to be concerned that other factors in the experiment (the presence or absence of some other element in the pathogenic process) are more likely to be responsible for the different behaviors of different variants than the variants themselves. This method is also subject to the limitation that it is difficult to measure the function of a protein variant when the function of the normal protein is largely or completely unknown.
A third approach is to use extensive prior knowledge of a protein's structure and function to predict the functional effect of a mutation on the protein. For example, the structure and function of a few proteins that are important to vision (e.g. rhodopsin) are exceedingly well worked out [14-17]. High resolution x-ray crystallographic data [18, 19] coupled with functional data from many experiments in several model organisms [20-22] allow one to infer a pathogenic effect of certain mutations. For example, any alteration of the residue (lysine 296) at which 11-cis retinal covalently attaches to the protein could be reasonably predicted to alter its function. This type of information can be used to contribute to the EPP value in an unbiased way by selecting a set of critical residues whose alteration would seem likely to affect the protein's function without first looking at a set of sequence variation data from humans. When so collected, the predicted functional information provides an independent piece of information about the possible effect of a sequence variation on the protein. The main limitation to this third approach at the present time is that this type of structural and functional information is available for only a small subset of potentially disease-causing genes, and usually only a small subset of residues within these genes. It is also prone to bias and to circular arguments (for example if one predicts a certain variant to cause disease after observing it in an affected patient).
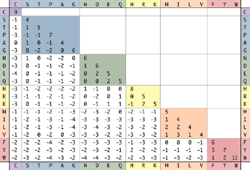
Blosum 62 Matrix
The final approach, and the one that is relied on most heavily by the EPP system, is to use evolutionary evidence gathered from thousands of proteins to assess the functional affect of a given change. This method will be discussed in detail below, but for the present purpose it is sufficient to say that every possible amino acid variation is assigned a value (B) from -4 to +3 in a table known as the blosum 62 substitution matrix [23]. B values below zero indicate amino acid changes that are more likely to have a functional effect than values of zero and above.
Association data used in the calculation
In addition to these four kinds of functional data, the EPP system takes three kinds of association data into account. The first is simply the difference in allele frequencies between patients and controls. That is, any variation that is proposed to cause a disease should be more common in patients than controls, but as will be shown below, such a skew by itself does not always reliably infer pathogenic potential. How much rarer does one expect a highly penetrant disease-causing variation to be in unaffected control individuals than in affected patients? The answer depends upon the prevalence and the presumed mode of inheritance of the disease in question. If a heterozygous sequence variation causes detectable disease at an early age in 95 percent of people who harbor it (i.e., is highly penetrant), one would expect the variation to be 19-fold less common in the general population than in the affected population. So, for a rare disease like retinitis pigmentosa (which occurs in about one in 4000 people) one would expect to randomly encounter an unaffected person with a true disease-causing mutation only once in 76,000 control samples – that is to say, almost never. In contrast, for an autosomal recessive condition in which two different disease alleles must be inherited in order for a person to manifest the disease, true disease-causing alleles are surprisingly common in the general population. For a 1 in 10,000 condition like Stargardt disease, one would expect true disease alleles to be present in about 1 in 50 people. If there are multiple different disease-causing mutations in a single disease gene, then the sum of these will be present in 1 in 50 people. Most people affected with X-linked disease are males with only one X chromosome and as a result the relationship between the disease prevalence and the allele frequency in the unaffected male population is very similar to the situation for autosomal dominant disease. That is, highly penetrant alleles that cause very rare X-linked diseases will be extremely rare (for all practical purposes zero) in the normal male population. The difference in the way that disease allele frequencies are related to disease prevalence is the factor that gives rise to the need for a different EPP calculation for dominant and recessive disease. For a rare dominant disease, any presence of a putative allele in the control group (barring diagnostic error or a sample swap) leads to an estimated pathogenic potential of zero. In contrast, a true highly penetrant allele for a rare recessive disease could easily be observed in a control group of 200 individuals and a low penetrance allele might be present in as many as a few percent of the general population (see Webster, et al. [13], for additional discussion).
A second kind of association data that can be used in the calculation of EPP is formal LOD score and haplotype analyses of large families. In this type of analysis, data from many genetic markers are considered, which allows one to detect things like ancestral relationships among families [24-26] and linkage disequilibrium among different variants in the same gene. These kinds of data should definitely be used on the "association side" of the EPP calculation when they exist. However, as with some of the types of functional data, this type of formal association information is available for only a small fraction of all sequence variants that are observed and cannot be relied upon for determining most EPP values. In practice, haplotype data are more likely to call a variant into question (e.g., a variant that is in clear disequilibrium with a more believable one) than it is to strengthen the argument for its pathogenic probability.
For autosomal dominant disease, the approach that we use most commonly for tabulating association information is a simplification of the LOD score method that simply counts the number of times a sequence variant has been observed to properly segregate with disease. This "M number" method will be described more fully below. However, for the purpose of understanding the EPP calculation, it is sufficient to know that an M number of 7 or higher is indicative of a less than 1 percent chance that the sequence variation and the disease phenotype are cosegregating by chance.
For rare recessive disease, when data are sufficient to suggest a statistically significant likelihood that the allele is more than 100 fold rarer in the control group than the disease group, the variant gets a supportive "point" toward being considered a highly penetrant allele. Conversely, when the data indicate that an allele is too common (as predicted by the Hardy-Weinberg equation – see glossary in Appendix) to be a highly penetrant allele, it loses "points" and is placed into the "possible low penetrance allele" (EPP=1) category.
Autosomal Dominant EPP Values
The EPP for autosomal dominant disease is calculated in the following way.
- nucleotide sequence variants that do not alter an amino acid (i.e. synonymous codon changes) and/or are present in approximately equal numbers in patients and controls. This category also contains rare variants that have been experimentally shown to have no effect on the function of the protein, variants whose disease association has been convincingly proven to be secondary to linkage disequilibrium with a more convincing mutation, and variants that have failed to segregate in affected individuals with rare diseases (e.g., Leu45Phe – see below).
- variants that alter an amino acid residue and are more common in patients than controls (small presence in controls is tolerable only for common disorders like AMD or glaucoma – that is, it is related to prevalence) but have no additional functional or association data to contribute to an argument that they are pathogenic.
- variants that alter an amino acid residue, are more common in patients than controls, and that exhibit either functional (B<0) or association (M>7) evidence for above average pathogenic potential.
- variants that alter an amino acid residue, are more common in patients than controls, and that exhibit both functional (B<0) and association (M>7) evidence for above average pathogenic potential.
For categories 2 and 3, specific functional data can be used instead of the B number when available for awarding the functional "point". Also, frameshift mutations, nonsense mutations (stops), multi-residue insertions or deletions, and mutations involving canonical splice sites are all awarded the functional "point" (blosum 62 calculations are only relevant to single amino acid changes).
Autosomal Recessive EPP Values
The EPP for autosomal recessive disease is calculated somewhat differently so that the numbers 0-3 will have a similar meaning to clinicians regardless of the inheritance pattern.
- nucleotide sequence variants that do not alter an amino acid (i.e. synonymous codon changes) and/or are present in approximately equal numbers in patients and controls. This category also contains rare variants that have been experimentally shown to have no effect on the function of the protein and variants whose disease association has been convincingly proven to be secondary to linkage disequilibrium with a more convincing mutation.
- variants that alter an amino acid residue and are more common in patients than controls but that are too common according to the Hardy-Weinberg equation to represent highly penetrant alleles.
- variants that alter an amino acid residue but are so rare that there is insufficient data to judge that they are either too common to be highly penetrant alleles or 100 times more common in patients than controls -- and -- which exhibit no functional evidence for above average pathogenic potential (i.e., B>-1).
- variants that alter an amino acid residue, are compatible with high penetrance from a Hardy-Weinberg perspective -- and -- exhibit one or more of the following characteristics: more than 100-fold more common in patients than controls, exhibit functional (B<0) evidence for above average pathogenic potential, or results in a frameshift, mis-splicing, or premature termination of translation.
When two affected siblings fail to share genotypes at a locus, it is concluded that that locus cannot be involved in their disease in a recessive Mendelian way and any observations of putative disease alleles are considered to have been made in control individuals. Such an observation could result in the demotion of a variant that was previously considered to be an EPP=3 because of a 100 fold or greater concentration in patients versus controls.